fol_Datos <- "../Datos/"
fol_Results <- "../Resultados/"1 Utilizar filtros simples
1.1 Ejercicio
- Crear una matriz de abundancia con únicamente los sitios de referencia de las hydro-eco-región de primer nivel: 2, 6 y 7 (Sur-Este de Francia)
- la matriz no puede tener especies que no están presentes en esos sitios
- ordenar las columnas por orden alfabético de los códigos de especies
- Comparar la riqueza de las especies en los 5% de los sitios de agua más acida y lo 5% de los sitios de agua más alcalina de esa nueva matriz
1.2 Parte 1: filtrar la matriz de diatomeas
# Aqui empiezan los codigos del ejercicio 1 sobre los filtros simples----
(load(paste0(fol_Datos,"diatomeas.RData")))[1] "sp_abund_mat" "reference_sites" "env_info"
[4] "coord_x_y" La manera más simple para filtrar las HER 2, 6 y 7 es utilizar este filtro
fil_her <- env_info$HER1Lyon == 2 | env_info$HER1Lyon == 6 | env_info$HER1Lyon == 7Note: existe una forma más simple que consiste en utilizar el operador %in%
fil_her <- env_info %in% c(2,6,7) El filtro completo para las HER y los sitios de referencia es:
fil_comp <- fil_her & reference_sitesPara conocer el numero de sitios que combinan las condiciones de HER y referencias, se puede utilizar la función sum, por el hecho que los vectores logicos almacenen sus contenidos utilizando los valores 0 y 1:
sum(fil_comp)[1] 36Ahora lo podemos aplicar a la matriz:
sp_abund_mat_filtered <- sp_abund_mat[fil_comp,]La función colSums nos permite conocer el total de abundancia de las taxones:
tot_abund_fil <- colSums(sp_abund_mat_filtered)
tot_abund_fil[1:10] ABRT ACOP ADDA ADMI ADMS ADPY ADSH ADSO ADSU AFOR
0 0 0 11503 14 7026 0 0 387 2 Como lo pueden ver en los 10 primeros taxones, con la matriz reducida, muchos taxones ya no tienen ningunos individuos.
Podemos utilizar la función as.logical para filtrar esas columnas: cuando la utilizamos con un vector numérico, da “FALSE” para todos los elementos que valen 0, y “TRUE” para los elementos superiores a 0.
as.logical(tot_abund_fil[1:10]) [1] FALSE FALSE FALSE TRUE TRUE TRUE FALSE FALSE TRUE TRUEAsí, se puede utilizar para guardar en la matriz unicamente las especies que tienen al menos un individuo:
dim(sp_abund_mat_filtered)[1] 36 926sp_abund_mat_filtered <- sp_abund_mat_filtered[, as.logical(colSums(sp_abund_mat_filtered))]
dim(sp_abund_mat_filtered)[1] 36 156Para ordenar las columnas por orden alfabetico, una de las soluciones es utilizar la función sort sobre los nombres de columnas (una alternativa sería utilizar la función order)
colnames(sp_abund_mat_filtered)[1:10] [1] "ADMI" "ADMS" "ADPY" "ADSU" "AFOR" "APED" "BVIT" "CAFF" "CHEL"
[10] "CPED" [1] "ACAF" "ADLA" "ADMI" "ADMS" "ADPY" "ADSU" "AEXM" "AFOR" "AINA"
[10] "AMUS"1.3 Parte 2: comparar la riqueza de los sitios acidos y alcalinos
Primero, tenemos que obtener el data.frame env_info filtrado como la matriz de abundancia:
env_info_filtered <- env_info[fil_comp,]Luego, tenemos que obtener una matriz que nos permita calcular la riqueza de los sitios. Otra vez, podemos utilizas las funciones de transformación entre modos numéricos y logicos. Eso nos permite obtener una matriz que tenga 0 (FALSE) cuando el taxon no está presente, y 1 (TRUE) cuando el taxon es presente.
sp_01_filtered <- sp_abund_mat_filtered
mode(sp_01_filtered) <- "logical"Así, calcular la riqueza se puede hacer con:
(riqueza <- rowSums(sp_01_filtered))662 680 682 684 692 695 723 735 736 746 747 748 749 750 751 752 753
21 23 13 17 13 23 14 11 12 28 10 16 13 13 13 18 11
754 755 757 758 759 760 761 762 763 764 789 795 796 798 800 802 814
12 15 16 11 5 12 11 11 15 13 27 30 15 23 21 16 9
815 818
39 48 Now, cual es el valor limite de las muestras más acidas y más alcalinas:
(acid5pc <- quantile(env_info_filtered$PH,0.05)) 5%
7.45 (alka5pc <- quantile(env_info_filtered$PH,0.95))95%
8.5 Ahora, cuales son las riquezas que corresponden a cada uno de esos grupos
(riq_acid <- riqueza[env_info_filtered$PH <= acid5pc])692 798
13 23 (riq_alka <- riqueza[env_info_filtered$PH >= alka5pc])682 684 747 750 761
13 17 10 13 11 Para guardar los resultados podemos utilizar:
2 Relaciones entre tablas en R
2.1 Ejercicio
La base de datos de diatomeas descrita en el primer ejercicio referencia los taxones por sus códigos, que están definidos por varios expertos europeos, pero esos códigos no son siempre especies, pueden corresponder a varios niveles taxonómicos.
Imaginemos que un grupo de investigadores quiere desarrollar un indice simple de calidad del agua como un cociente entre la abundancia de la familia Fragilariaceae y la familia Naviculaceae (en realidad no tiene mucho sentido, es más un ejercicio de programación que realmente un indíce que tenga sentido).
- Calcular ese indice para cada una de las muestras de la matriz sp_abund_mat
- Comparar los valores entre los sitios de referencia y los demás
Ejercicio suplementario para utilizar el mismo tipo de razonamiento (solo para si tenemos tiempo):
¡Hacer una matriz por nivel taxonomico!
2.2 Parte 1: Indice Fragilariacea/Naviculaceae
Lo que hacemos es buscar la correspondancia entre los colnames de la matriz de abundancia de las diatomeas:
Luego miramos cuales son las columnas que corresponden a cada una de las familias del indice:
Finalmente calculamos los rowSums de las matriz filtradas con cada una de esas familias:
Finalmente hacemos un grafico boxplot del indice en función de los sitios de referencia (el signo ~ quiere decir en función de)
boxplot(indexFraNav~reference_sites)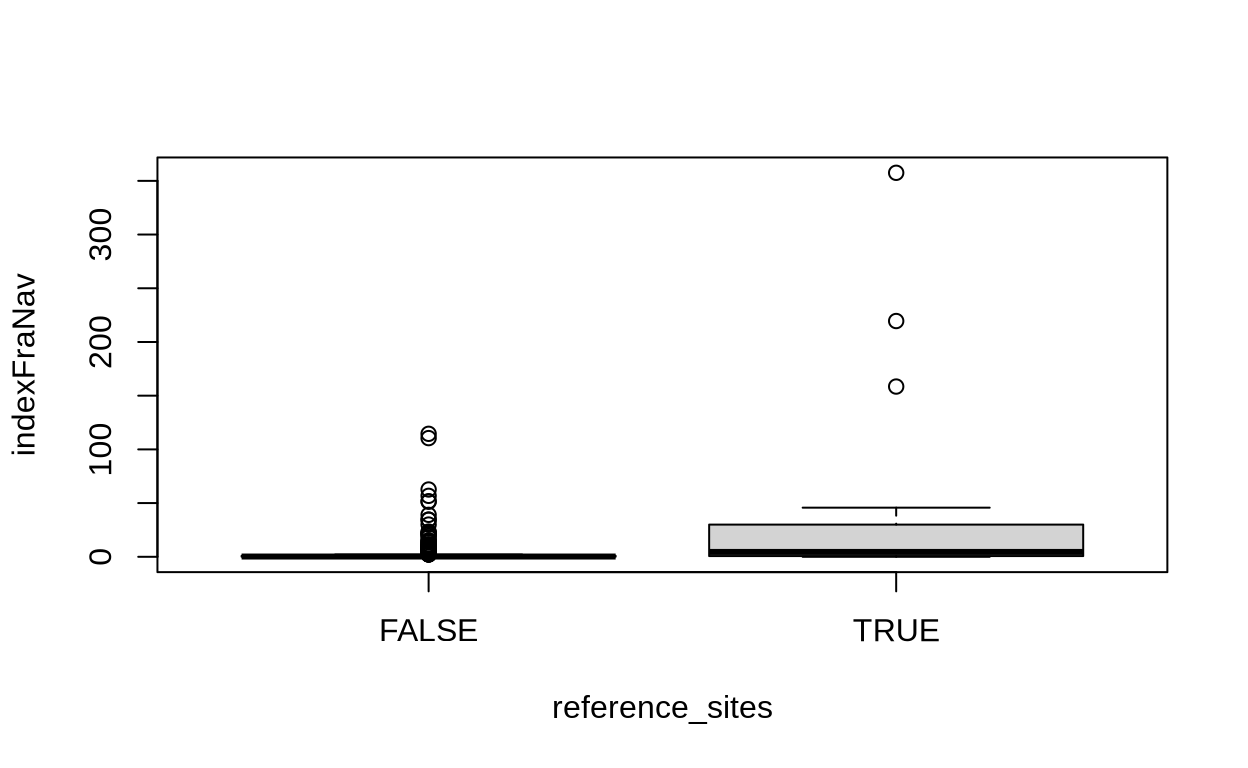
2.3 Parte 2: Matriz por familia
# Buscamos, para cada taxón, la familia que corresponde
family <- taxonomy$family[m_sp_mat_taxo]
# Miramos la lista de familia (unica) y excluímos los taxones que no tienen una información de familia
un_family <- na.omit(unique(family))
# Creamos la matriz
mat_diat_fam <- matrix(NA, nrow=nrow(sp_abund_mat), ncol=length(un_family),dimnames=list(NULL,un_family))
# Hacemos un bucle que toma cada familia una por una
# Para cada familia filtramos la matriz de abundancia, y calculamos la suma por fila
for(i in un_family)
{
#Caso 1: solo hay un taxon en la familia
if(sum(family==i,na.rm=T)==1)
{mat_diat_fam[,i] <- sp_abund_mat[,which(family==i)]
}else{ # Caso 2 existe una matriz de más de una columna en esta familia
mat_diat_fam[,i]<-rowSums(sp_abund_mat[,which(family==i)])
}
}3 Leer un archivo complejo
3.1 Ejercicio
Pasar los datos contenidos en el archivo “fitosociologiaFagusFrancia.xlsx” en:
- Una matriz de cobertura sitio x especie y la cobertura en porcentaje en la matriz
- Un data frame de información sobre los sitios
No modifiquen el archivo excel, todo se puede hacer en R, es por enfrentarse a este tipo de dificultades que nos mejoremos en R.
3.2 Parte 1: Leer la información desde el archivo Excel
Existen varias estrategías para resolver este ejercicio, no descarten una solución por ser diferente de la solución que yo proponga…
Es muy probable que tengamos que leer cada una de las pestañas.
En esos casos, lo más practico es utilizar las funciones loadWorkbook
# Ejercicio 2 sobre la lectura de archivos complejos ----
require(openxlsx)
fFF <- loadWorkbook(paste0(fol_Datos,"fitosociologiaFagusFrancia.xlsx"))
(sheetNames<-names(fFF))[1] "Lapraz 1963 E2M" "Lapraz 1963 E2M 3"Lo que vamos a hacer es leer las 2 pestañas de manera totalmente bruta y ponerlas en una lista, después, podemos extraer la información de esa lista. Vamos a utilizar los argumentos colNames=F y rowNames=F, para poder acceder a todas la información desde los elementos de la lista, incluído las filas y columnas que vamos a utilizar como nombres. Vamos a utilizar los argumentos skipEmptyRows=T y skipEmptyCols=T para suprimir automaticamente las columnas y filas vacías.
Ahora, vamos a suprimir la ultima columna, que tiene información que no hace parte de la tabla
3.3 Parte 2: información ambiental
Determinamos el número de fila hasta donde va la información ambiental. En las 2 pestañas, es la fila antes de que la columna 1 contenga “Genre” (genero, en francés).
(lastRowEnv_1 <- which(rawList[[1]][,1] == "Genre")-1)[1] 6(lastRowEnv_2 <- which(rawList[[2]][,1] == "Genre")-1)[1] 6Ahora podemos extraer los datos:
- los nombres de los relevés (muestras fitosociologicas) están en la primera fila (pero son iguales en las dos pestañas, vamos a tener que modificarlos para poder asociar las 2 tablas)
- la segunda columna tiene que estar suprimida porque no tiene datos
- queremos que las filas sean las unidades de muestreo y las columnas las variables: esa operación se llama “transponer” y se hace con la función
t. Nota: acordarse que los data.frame solo tienen sentido cuando las variables están en columnas: las columnas son vectores que tienen un modo.
# Extraer las filas que corresponden, las columnas que tienen un numero de relevé,
# sacar la columna que contiene los nombres de filas, transponer lo extraído y
# pasarlos en formato data.frame
fFF_env_info1 <- data.frame(t(rawList[[1]][1:lastRowEnv_1, !is.na(rawList[[1]][1,])][,-1]))
fFF_env_info2 <- data.frame(t(rawList[[2]][1:lastRowEnv_2, !is.na(rawList[[2]][1,])][,-1]))
# Añadir los colnames
colnames(fFF_env_info1) <- rawList[[1]][1:lastRowEnv_1,1]
colnames(fFF_env_info2) <- rawList[[2]][1:lastRowEnv_2,1] Ahora tenemos que manejar los modos de las columnas, el problema es que la variable “Taux de carbonate” (Tasa de carbonatos en francés) tiene el valor “Traces” cuando hay carbonatos, pero bajo el umbral de cuantificación. Es siempre un problema complicado en las bases de datos ambientales, pero, por ahora, podemos ponerlos el valor “0”.
La función type.convert permite reiniciar los modos de las columnas de un data.frame.
Es la función que llama read.table para determinar los tipos de datos según los contenidos de los archivos.
fFF_env_info1$`Taux de carbonates`[fFF_env_info1$`Taux de carbonates` == "Traces"] <- 0
fFF_env_info1 <- type.convert(fFF_env_info1,as.is=T)
fFF_env_info2 <- type.convert(fFF_env_info2,as.is=T)Tenemos que dar un nombre a cada una de las unidades de muestreo, para que no haya conflictos al momento de juntar las tablas.
Lo que les propongo es juntar un prefijo “LZ1_” o “LZ2_” con el número de relevé.
La función paste nos permite hacer eso:
Ahora tenemos que juntar las tablas. Para eso tenemos que determinar las columnas que existen solo en una de las tablas.
# Cuales son todos los nombres de columnas
totCol <- union(colnames(fFF_env_info1),colnames(fFF_env_info2))
# Cuales las columnas que faltan en la tabla1
(missingCol1 <- totCol[! totCol %in% colnames(fFF_env_info1)])[1] "Surface (m²)"# Cuales las columnas que faltan en la tabla2
(missingCol2 <- totCol[! totCol %in% colnames(fFF_env_info2)])[1] "Taux de carbonates"# Crear las columnas faltantes y llenarlas de NA
fFF_env_info1[,missingCol1]<-NA
fFF_env_info2[,missingCol2]<-NA
# Hacer que las columnas de las dos tablas tengan el mismo orden
fFF_env_info1<-fFF_env_info1[totCol]
fFF_env_info2<-fFF_env_info2[totCol]Ahora sí, podemos juntar las tablas con la función rbind (concatenar por filas)
fFF_env_info<-rbind(fFF_env_info1, fFF_env_info2)3.4 Parte 3: matrices de especies
Lo que nos toca extraer para obtener las matrices es:
- la filas desde lastRowEnv_1 + 2
- las columnas desde la columna 3
- pasar en formato matriz con la función
as.matrix - transponer el resultado para tener los taxones como columnas
Cuales son los nombres completos de los taxones, aqui tambien utilizamos la función paste
(tax1 <- paste(
rawList[[1]][(lastRowEnv_1+2):nrow(rawList[[1]]),1],
rawList[[1]][(lastRowEnv_1+2):nrow(rawList[[1]]),2]
))[1:10] [1] "Carpinus betulus" "Quercus robur"
[3] "Quercus petraea" "Viburnum lantanta"
[5] "Mercurialis perennis" "Conopodium denudatum"
[7] "Viola hirta" "Helleborus viridis"
[9] "Lathyrus latifolius" "Viburnum tinus" (tax2 <- paste(
rawList[[2]][(lastRowEnv_2+2):nrow(rawList[[2]]),1],
rawList[[2]][(lastRowEnv_2+2):nrow(rawList[[2]]),2]
))[1:10] [1] "Castanea sativa" "Quercus robur"
[3] "Carpinus betulus" "Quercus toza"
[5] "Populus tremula" "Quercus petraea"
[7] "Lonicera periclymenum" "Teucrium scorodonia"
[9] "Rhamnus frangula" "Viola riviniana" Pongamos los “cd_rel” como rownames de las matrices
Ahora vamos a mirar cuales son las especies que están solo en una matriz, y pegar una matriz de NA con las especies faltantes en cada caso, utilizando las funciones matrix y cbind.
# Lista completa de especies
totSp <- union(colnames(raw_mat1), colnames(raw_mat2))
# especies faltantes en cada matriz
missingSpMat1 <- totSp[! totSp %in% colnames(raw_mat1)]
missingSpMat2 <- totSp[! totSp %in% colnames(raw_mat2)]
# crear las matrices de NA con las especies faltantes.
# Anotar: el argumento dimnames tiene que ser una lista: el primer elemento contiene los
# nombres de filas, el segundo los nombres de columnas
matSupp1 <- matrix(NA, nrow=nrow(raw_mat1), ncol=length(missingSpMat1),
dimnames=list(rownames(raw_mat1), missingSpMat1))
matSupp2 <- matrix(NA, nrow=nrow(raw_mat2), ncol=length(missingSpMat2),
dimnames=list(rownames(raw_mat2), missingSpMat2))
# Ahora pegamos por columna esas matrices a las matrices originales
raw_mat1 <- cbind(raw_mat1, matSupp1)
raw_mat2 <- cbind(raw_mat2, matSupp2)
#Ponemos las columnas en el mismo orden en las 2 matrices
raw_mat1 <- raw_mat1[,totSp]
raw_mat2 <- raw_mat2[,totSp]Ahora juntamos las matrices
raw_mat <- rbind(raw_mat1, raw_mat2)3.5 Parte 4: cambiar de la escala de Braun-Blanquet a los valores medianos de cobertura
Copiamos la tabla de correspondencia desde la información del ejercicio:
bbScale <-data.frame(
code=c("r","+",as.character(1:5)),
description= c("Menos del 1% de cobertura, 3-5 individuos", "Menos del 5% de cobertura, pocos individuos","~5% más individuos","5%-25%","25%-50%","50%-75%","75%-100%"),
minPercent=c(0,1,5,5,25,50,75),
maxPercent=c(1,5,5,25,50,75,100)
)
bbScale$finalVal <- (bbScale$minPercent+bbScale$maxPercent)/2La función match nos permite hacer la correspondencia entre la matriz total y la tabla bbScale.
Vamos a utilizar una propiedad de las matrices: ¡son vectores con una estructura de filas y columnas!
Así, podemos utilizar directamente la función match para determinar a que elementos de bbScale corresponden cada uno de los elementos de la matriz, uno por uno.
m_raw_mat_code <- match(raw_mat,bbScale$code)¡Ahora, simplemente creamos la matriz final con los valores “finalVal” en lugar de los code!
Reemplazamos los valores NA por 0:
matCobertura[is.na(matCobertura)] <- 0Pueden mirar el resultado en RStudio utilizando View(matCobertura)
Lo que voy a hacer ahora es exportar esas tablas en un archivo excel:
# Crear el workbook
wbFinal <- createWorkbook()
# Crear las pestañas
addWorksheet(wbFinal,"Sampling units")
addWorksheet(wbFinal,"Cobertura")
# Poner los datos
writeData(wbFinal,"Sampling units", fFF_env_info, rowNames = F)
writeData(wbFinal,"Cobertura", matCobertura, rowNames = T)
# Exportar el archivo excel
saveWorkbook(wbFinal, file = paste(fol_Results,"fitosociologíaFagusFranciaFinal.xlsx"), overwrite = T)4 Construir funciones
4.1 Ejercicio
Contruir las funciones en R que permiten pasar de un formato “base de datos” a una matriz de abundancia o de presencia.
Las funciones deben tener los argumentos adecuados para :
- poder dar el nombre de la columna que contiene los taxones y de las unidades de muestreo
- poder manejar casos de presencia/ausencia, abundancia
- poder llenar las matrices con 0 o NA, y transformar las matrices con NA o 0 cuando las especies son ausentes
- añadir todas las opciones que les parecen útil (un ejercicio más tarde consistira en publicar esas funciones en la forma de un paquete R en GitHub)
Construir una función que permita utilizar el formato “base de datos” y hacer la suma de todos las filas que tienen el mismo taxon en la misma unidad de muestreo, y utilizarla para construir la matriz de genero de diatomeas (ver ejercicio previo).
4.2 Parte 1: crear las funciones
4.2.1 Pasar del formato base de datos al formato matriz
(load("../Datos/bog_chinga.RData"))[1] "df_bog_chinga" "mat_bog_chinga"df_bog_chinga UniMuestreo Especie abundancia
1 Bogotá Canis lupus 3000
2 Bogotá Felis catus 1000
3 Chingaza Tremarctos ornatus 50
4 Chingaza Canis lupus 100
5 Chingaza Odocoileus virginianus 200Para construir la función, vamos a utilizar el principio de which con arr.ind=T.
En https://marbotte.github.io/cursoBiodata/doc_R_buscarRelacionar.html#arrays podemos ver eso:
4.2.1.1 Principio director
load("../Datos/bog_chinga.RData")
mat_bog_chinga Canis lupus Felis catus Tramarctos ornatus
Bogotá 3000 1000 0
Chingaza 100 0 50
Odocoileus viginianus
Bogotá 0
Chingaza 200(sup0 <- which(mat_bog_chinga > 0, arr.ind = T)) row col
Bogotá 1 1
Chingaza 2 1
Bogotá 1 2
Chingaza 2 3
Chingaza 2 4mat_bog_chinga[sup0][1] 3000 100 1000 50 200Vamos utilizar el mismo principio, pero al revez: vamos a utilizar una matriz que contiene las columnas “row” y “col” para dar las “direcciones” en la matriz que hay que llenar
4.2.1.2 Hacerlo fuera de una función
Lo primero que tenemos que hacer es tener la lista de columnas y filas para la matriz, para df_bog_chinza, es:
(ROW <- unique(df_bog_chinga$UniMuestreo))[1] "Bogotá" "Chingaza"(COL <- unique(df_bog_chinga$Especie)) [1] "Canis lupus" "Felis catus"
[3] "Tremarctos ornatus" "Odocoileus virginianus"Ahora que tenemos eso, podemos crear una matriz llena de 0 con los nombres de taxones y unidades de muestreo:
Canis lupus Felis catus Tremarctos ornatus
Bogotá 0 0 0
Chingaza 0 0 0
Odocoileus virginianus
Bogotá 0
Chingaza 0Ahora hacemos la matriz con row y col, por ejemplo para los rows:
Entonces la matrice es:Así podemos llenar la matriz con esas “direcciones”:
mat[matRowCol]<-df_bog_chinga$abundancia
mat Canis lupus Felis catus Tremarctos ornatus
Bogotá 3000 1000 0
Chingaza 100 0 50
Odocoileus virginianus
Bogotá 0
Chingaza 2004.2.1.3 Programar la función
dbTab2mat <-
function(dbTab,col_samplingUnits="SU",col_species="sp",col_content="abundance",empty=NA,checklist=F)
{
COLS<-unique(as.character(dbTab[,col_species]))
ROWS<-unique(as.character(dbTab[,col_samplingUnits]))
arr.which<-matrix(NA,ncol=2,nrow=nrow(dbTab),dimnames=list(1:nrow(dbTab),c("row","col")))
arr.which[,1]<-match(as.character(dbTab[,col_samplingUnits]),ROWS)
arr.which[,2]<-match(as.character(dbTab[,col_species]),COLS)
# Esta linea es para determinar el modo de los datos, según los argumentos
# Si checklist está verdadero, entonces el modo es TRUE/FALSE (logico), sino corresponde al modo de la columna col_content
modeContent<-ifelse(checklist,"logical",mode(dbTab[,col_content]))
# Ahora que tenemos el modo, entonces podemos saber con que llenar la matriz:
if(is.na(empty)){empty<-switch(modeContent,character="",numeric=0,logical=F)}
# Creamos la matriz
res<-matrix(empty,ncol=length(COLS),nrow=length(ROWS),dimnames=list(ROWS,COLS))
# La llenamos
if(checklist){ res[arr.which]<-T}else{res[arr.which]<-dbTab[,col_content]}
return(res)
}4.2.1.4 Aplicar la función
dbTab2mat(df_bog_chinga, col_samplingUnits = "UniMuestreo", col_species = "Especie", col_content = "abundancia") Canis lupus Felis catus Tremarctos ornatus
Bogotá 3000 1000 0
Chingaza 100 0 50
Odocoileus virginianus
Bogotá 0
Chingaza 2004.2.2 Para pasar de matriz a formato de base de datos
4.2.2.1 Principio director
Vamos a utilizar which con arr.ind=T para determinar cuales elementos de la matrice corresponden a una abundancia superior a 0
which(mat_bog_chinga>0,arr.ind =T) row col
Bogotá 1 1
Chingaza 2 1
Bogotá 1 2
Chingaza 2 3
Chingaza 2 4Desde eso, ¡solo nos queda reemplazar los contenidos numericos de esa matriz por los nombres de linea y columna la matriz!
4.2.3 Hacer la función
mat2dbTab <-
function(mat,checklist=F,col_samplingUnits="SU", col_taxon="taxon",col_content="Abundance")
{
#busquemos los contenidos de la matriz superiores a 0
W<-which(mat>0,arr.ind=T)
if(!checklist){# Si es presencia ausencia
dbTab<-data.frame(SU=rownames(mat)[W[,"row"]],sp=colnames(mat)[W[,"col"]],ab=mat[W])
}else{# Si no es presencia ausencia
dbTab<-data.frame(SU=rownames(mat)[W[,"row"]],sp=colnames(mat)[W[,"col"]])
}
# Reorganizamos la tabla por sitios, luego por especie
dbTab<-dbTab[order(dbTab$SU,dbTab$sp),]
# Ponemos los colnames
COLNAMES <- c(col_samplingUnits,col_taxon)
if(!checklist){COLNAMES<-c(COLNAMES,col_content)}
colnames(dbTab)<-COLNAMES
return(dbTab)
}4.2.4 Aplicar la función
mat2dbTab(mat_bog_chinga) SU taxon Abundance
1 Bogotá Canis lupus 3000
3 Bogotá Felis catus 1000
2 Chingaza Canis lupus 100
5 Chingaza Odocoileus viginianus 200
4 Chingaza Tramarctos ornatus 50mat2dbTab(mat_bog_chinga,checklist = T) SU taxon
1 Bogotá Canis lupus
3 Bogotá Felis catus
2 Chingaza Canis lupus
5 Chingaza Odocoileus viginianus
4 Chingaza Tramarctos ornatus4.3 Parte 2: una función para sumar los casos repetidos
Imaginamos que añadimos encontremos más gatos (50) en Bogotá:
(df_bog_chinga<-rbind.data.frame(df_bog_chinga,
data.frame(UniMuestreo="Bogotá",
Especie="Felis catus",
abundancia=50
))) UniMuestreo Especie abundancia
1 Bogotá Canis lupus 3000
2 Bogotá Felis catus 1000
3 Chingaza Tremarctos ornatus 50
4 Chingaza Canis lupus 100
5 Chingaza Odocoileus virginianus 200
6 Bogotá Felis catus 50Ahora tenemos más de una vez la asociación entre “Bogotá” y “Felis catus”, tenemos que sumarlas:
Vamos a utilizar la función by
Ahora utilizamos Reduce para transformar la lista en un data.frame
(gatosMas <- Reduce(rbind,resultSUM)) UniMuestreo Especie abundancia
1 Bogotá Canis lupus 3000
4 Chingaza Canis lupus 100
2 Bogotá Felis catus 1050
5 Chingaza Odocoileus virginianus 200
3 Chingaza Tremarctos ornatus 50Para hacer una función generica:
sumRepeated(df_bog_chinga, col_sampUnit = "UniMuestreo", col_taxon="Especie", col_content = "abundancia") UniMuestreo Especie abundancia
1 Bogotá Canis lupus 3000
4 Chingaza Canis lupus 100
2 Bogotá Felis catus 1050
5 Chingaza Odocoileus virginianus 200
3 Chingaza Tremarctos ornatus 504.4 Parte 3: utilizar las funciones para resolver el ejercicio de matriz de genero de diatomeas
Primero saquemos la matriz de diatomeas en forma de base de datos:
dbTabDiat <- mat2dbTab(sp_abund_mat)
head(dbTabDiat) SU taxon Abundance
1 1 ABRT 1
62 1 ACOP 1
314 1 ADDA 1
332 1 ADMI 270
1069 1 ADMS 1
1126 1 ADPY 27Ahora buscamos los generos:
SU taxon Abundance genus
1 1 ABRT 1 Achnanthidium
62 1 ACOP 1 Amphora
314 1 ADDA 1 Achnanthidium
332 1 ADMI 270 Achnanthidium
1069 1 ADMS 1 Adlafia
1126 1 ADPY 27 AchnanthidiumdbTabDiatGenus<-dbTabDiat[,-which(colnames(dbTabDiat)=="taxon")]
dbTabDiatGenus<-sumRepeated(dbTabDiatGenus, col_taxon="genus")
head(dbTabDiatGenus) SU Abundance genus
38585 109 2 Achnanthes
38726 120 2 Achnanthes
38727 127 1 Achnanthes
38586 137 2 Achnanthes
38587 151 2 Achnanthes
38994 166 2 AchnanthesSolo nos queda volver a una matriz:
matDiatGenus<-dbTab2mat(dbTabDiatGenus,col_samplingUnits = "SU",col_species = "genus",col_content = "Abundance")4.5 Exportar